Что такое клинические испытания. Как проходят фазы клинических испытаний. Зачем нужны клинические испытания в медицине. Какие бывают типы клинических исследований. Как проводятся доклинические испытания лекарств.
Что такое клинические испытания и зачем они нужны
Клинические испытания — это исследования для тестирования новых лекарств, устройств или других методов лечения на людях-добровольцах. Их главная цель — проверить эффективность и безопасность новых медицинских разработок перед их широким внедрением в практику.
Основные задачи клинических испытаний:
- Определить, работает ли новое лечение у людей
- Оценить, насколько оно эффективно по сравнению с существующими методами
- Выявить возможные побочные эффекты и риски
- Определить оптимальные дозы и схемы применения
Клинические испытания могут занимать годы, но они необходимы, чтобы убедиться в пользе и безопасности новых методов лечения, прежде чем применять их широко.

Типы и этапы клинических исследований
Клинические испытания обычно проходят в несколько этапов или фаз:
Доклинические (лабораторные) исследования
Это первый этап, когда новое вещество или метод тестируется в лаборатории:
- На культурах клеток
- На лабораторных животных
Цель — получить предварительные данные о безопасности и эффективности, прежде чем переходить к испытаниям на людях.
Фаза 0
Это очень ранние исследования на небольшом числе людей (до 15 человек). Цели:
- Проверить, как лекарство ведет себя в организме человека
- Оценить, достигает ли оно нужных тканей
- Определить, как на него реагируют клетки
Используются очень низкие дозы, поэтому риск для участников минимален.
Фаза I клинических испытаний: оценка безопасности
Это первые полноценные исследования на людях. Основные задачи фазы I:
- Определить безопасную дозу препарата
- Выявить побочные эффекты
- Понять, как лекарство ведет себя в организме
Ключевые особенности исследований фазы I:
- Участвует небольшое число людей (до нескольких десятков)
- Начинают с очень низких доз, постепенно их повышая
- Ведется тщательное наблюдение за состоянием участников
- Эффективность не является основной целью
Хотя риски в фазе I выше, чем на других этапах, для некоторых пациентов с тяжелыми заболеваниями это может быть шансом получить новое перспективное лечение.

Фаза II клинических испытаний: оценка эффективности
На этом этапе проверяется, работает ли новый метод лечения при конкретных заболеваниях. Основные цели фазы II:
- Определить, эффективно ли лечение
- Оценить кратко- и среднесрочные побочные эффекты
- Уточнить оптимальные дозы и схемы применения
Особенности исследований фазы II:
- Участвует от 25 до 100 пациентов с определенным заболеванием
- Все получают одинаковую дозу, определенную в фазе I
- Могут сравниваться разные дозы или схемы лечения
- Не используется плацебо
Если лечение показывает хорошие результаты в фазе II, оно переходит в более масштабную фазу III.
Фаза III клинических испытаний: сравнение с существующими методами лечения
Это последний этап перед возможным одобрением нового метода лечения. Основные задачи:
- Сравнить эффективность нового лечения со стандартными методами
- Выявить редкие побочные эффекты
- Оценить долгосрочную безопасность
Ключевые особенности исследований фазы III:
- Участвуют сотни или тысячи пациентов
- Проводятся в разных медицинских центрах, часто международные
- Часто используется рандомизация и двойной слепой метод
- Может применяться плацебо (но не вместо стандартного лечения)
- Длятся дольше, чем исследования предыдущих фаз
Успешное прохождение фазы III — ключевое условие для одобрения нового лечения регулирующими органами.

Процесс одобрения новых лекарств
После завершения клинических испытаний разработчики подают заявку на одобрение нового препарата в регулирующие органы (в США — FDA). Процесс включает:
- Подача досье с результатами всех исследований
- Экспертная оценка данных об эффективности и безопасности
- Инспекции производственных площадок
- Решение об одобрении или отказе
При положительном решении новый препарат может выйти на рынок. Однако наблюдение за его безопасностью продолжается и после начала широкого применения.
Значение клинических испытаний для развития медицины
Клинические испытания играют ключевую роль в разработке новых методов лечения:
- Обеспечивают научно обоснованный подход к оценке новых препаратов
- Позволяют выявить неэффективные или небезопасные методы до их широкого внедрения
- Способствуют постоянному совершенствованию методов лечения
Благодаря клиническим исследованиям медицина постоянно развивается, появляются более эффективные и безопасные способы лечения многих заболеваний.

Как определить фазу и ноль мультиметром: инструкции, фото, видео
Чтобы правильно подключить приборы освещения, розетки и другие электроустройства нужно знать, где фаза и ноль. Для этого можно воспользоваться очень полезным и функциональным измерителем — мультиметром. Несмотря на кажущуюся простоту этого прибора, нужно научиться им пользоваться, в некоторых случаях одно неверное действие может привести к неприятным и даже плачевным результатам. Мы расскажем вам, как определить фазу и ноль мультиметром, и вы сможете безопасно организовать электричество в своём доме.
Contents
- 1 Для неискушённых пользователей: что такое фаза и ноль
- 2 Самое важное: правила безопасности
- 3 Как определить фазу мультиметром
- 4 Как найти ноль мультиметром
- 4.1 Вопрос — ответ
Для неискушённых пользователей: что такое фаза и ноль
Чтобы понять, как определить фазу и ноль мультиметром, нужно сначала узнать, что такое «фаза и ноль». Здесь нам пригодится элементарная физика. Вспомним определение электротока, знакомое многим из нас со школы, — это упорядоченное движение заряженных частиц, то есть электронов. Все электросети сгруппированы так:
Здесь нам пригодится элементарная физика. Вспомним определение электротока, знакомое многим из нас со школы, — это упорядоченное движение заряженных частиц, то есть электронов. Все электросети сгруппированы так:
- С постоянным током, когда частицы движутся в едином направлении.
- С переменным, когда направление носит переменчивый характер.
Нам нужен второй вид. Переменная сеть включает в себя две части:
- Фаза (официальное название — рабочая фаза), по которой идёт рабочее напряжение.
- Ноль или пустая фаза, необходимая для образования замкнутой сети, чтобы подключались и работали электроприборы. Кроме того, она используется для сетевого заземления.
Когда электроприборы включаются в однофазку, расположение этих двух фаз не имеет значения. Но для монтажа электропроводки и её присоединения к общедомовой сети без этих знаний не обойтись.
О том, как проверить мультиметром фазу и ноль, мы и поговорим далее, но сначала вспомним простейшие меры безопасности.
Самое важное: правила безопасности
- Не используйте нерабочие щупы.
- Не используйте измеритель там, где царит высокая влажность.
- При выборе диапазона измерений переключатель важно сразу ставить к наибольшему значению во избежание поломки мультиметра.
- Не изменяйте измерительные границы или режим тестера прямо в ходе замеров. Проще говоря, не вертите переключатель мультиметра, когда делаете измерение.
- Перед эксплуатацией мультиметра прочитайте руководство по его применению. Есть разные модели и обозначения. Чтобы правильно расставить щупы, выбрать точный режим и диапазон значений, изучите руководство к своей модели тестера. Полезно прочитать и наш материал о том, как пользоваться мультиметром.
Как определить фазу мультиметром
Для начала включите тестер и выберете функцию тестирования напряжения переменного тока. Чаще всего она отмечена знаком V~. Сразу ставим максимальный предел измерения, например, 750В. Не забудьте правильно установить щупы в гнезда.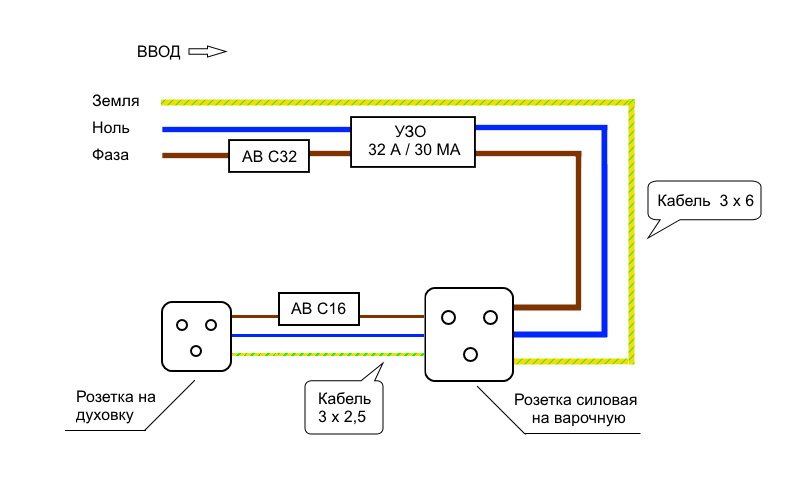 Обычно черный подключается к отверстию с надписью COM, а красный к VΩmA.
Обычно черный подключается к отверстию с надписью COM, а красный к VΩmA.
Кстати, если вы хотите убедиться в работоспособности определённого тестера (а это очень важно!), проверьте свою розетку. Сделать это очень просто: вставить щупы в розеточные гнёзда. О полярности не беспокойтесь, здесь она значения не имеет. Главное правило — не касайтесь руками частей щупов, которые проводят ток. Если с вашим тестером всё в порядке, нет затруднений с электроснабжением и подключением розетки, на дисплее вы увидите значение около 220-230В.
Теперь можно продолжить рассказывать о том, как найти мультиметром фазу в розетке 220В.
Проще всего обстоят дела, если перед нами три проводка: земля, ноль и фаза. Всё, что нужно сделать в такой ситуации — проверить напряжение всех пар. Между землей и нулём напряжения почти нет, значит, другой проводок — фаза.
Если же перед вами два проводка, всё немного иначе. Теперь нам нужно организовать подходящие условия для движения электричества по прибору. Итак, дальнейшие действия для проверки фазы мультиметром:
Итак, дальнейшие действия для проверки фазы мультиметром:
- Наконечником алого провода тестера дотрагиваемся до исследуемого проводка.
- Наконечник темного провода мультиметра прижимаем пальцами или касаемся им заземленного предмета (второй вариант предпочтительнее!). Им может быть стальной каркас рядом стоящей стены, отопительная батарея и т.п. Главное — выбрать заземленный предмет.
- Смотрим на показания мультиметра. Если вы видите показания, приближенные к 220В, значит, вы нашли фазу. Цифра может чуть отличаться в зависимости от условий, но будет находиться в пределах указанного значения. Если проверяемый вами кабель не является фазой, значит, вы увидите на дисплее 0 или немного вольт.
Есть ли риск в этом методе? Да, но он очень маленький. Дело в том, что сетевое напряжение движется через значительное сопротивление резистора, который встроен в наш измерительный прибор. Поэтому удара током нет. А рабочий этот резистор или нет, мы предварительно проверяем с помощью розетки способом, который описали выше. Без рабочего резистора, конечно, складываются отличные предпосылки для короткого замыкания, а его не заметить невозможно.
Без рабочего резистора, конечно, складываются отличные предпосылки для короткого замыкания, а его не заметить невозможно.
И лучше всего не зажимать наконечник пальцами, а использовать для этого заземлённые устройства. Но это возможно не всегда. Если вы будете использовать свою руку, советуем не пренебрегать такими принципами безопасности, как резиновый коврик под ногами или диэлектрические ботинки. Кроме того, прикоснитесь к щупу правой рукой сначала быстро: если нет никаких неприятных ощущений, то выполняйте измерения.
Рекомендуем посмотреть видео о том, как узнать мультиметром фазу и ноль:
Конечно, не забудьте перед описанными манипуляциями выбрать режим измерения именно напряжения переменного тока.
Если же вы не уверены, что всё пройдет благополучно, не беритесь за это дело, а доверьте опытным электрикам. Кроме того, можно использовать вместо мультиметра индикаторную отвертку (её индикатор загорается/не загорается при проверке).
А вот ещё одно интересное видео в тему, как мультиметром узнать, где фаза:
Как найти ноль мультиметром
Логично предположить, что ноль располагается по отношению к фазе, поэтому искать его легко: если вы нашли фазу, второй проводок из пары — ноль. Но не всё так просто, потому что другой провод может также быть землей. Ноль и заземление почти одинаковы. Иногда эти два провода связываются в щите и выявить их весьма нелегко. Как определить ноль мультиметром?
Советуется выключить кабель ввода от заземлительной шины в щитке. В таком варианте, когда будет проверяться напряжение между землёй и фазой, 220В не будет, как при тестировании ноля и фазы. Если в щитке имеется дифференциальная защитная система, она проявит себя, когда будут проверяться заземлительные проводки относительно иного проводника, даже если он нулевой.
В таком варианте, когда будет проверяться напряжение между землёй и фазой, 220В не будет, как при тестировании ноля и фазы. Если в щитке имеется дифференциальная защитная система, она проявит себя, когда будут проверяться заземлительные проводки относительно иного проводника, даже если он нулевой.
Как проверить ноль мультиметром в розетке:
- Красный провод мультиметра подвести к дырке, где фаза.
- Черный провод соединить сначала с одним контактом, потом с другим.
- Зафиксировать оба напряжения. Где оно меньше — там земля, где чуть больше — ноль.
Теперь вы знаете, как определить фазу и ноль мультиметром. Делитесь в комментариях своим опытом.
Желаем безопасных и точных измерений!
Вопрос — ответ
Вопрос: Как определить фазу цифровым мультиметром?
Имя: Кирилл
Ответ: Включите тестер и выберете функцию тестирования напряжения переменного тока. Чаще всего она отмечена знаком V~. Поставьте максимальный предел измерения, например, 750В. Не забудьте правильно установить щупы в гнезда. Обычно черный подключается к отверстию с надписью COM, а красный к VΩmA.
Поставьте максимальный предел измерения, например, 750В. Не забудьте правильно установить щупы в гнезда. Обычно черный подключается к отверстию с надписью COM, а красный к VΩmA.
Вопрос: Как безопасно найти фазу мультиметром?
Имя: Матвей
Ответ: Для этого нужно убедиться в работоспособности мультиметра с помощью проверки розетки. Вставьте щупы в розеточные гнёзда, не касайтесь руками частей щупов, которые проводят ток. Если с вашим тестером всё в порядке, нет затруднений с электроснабжением и подключением розетки, на дисплее вы увидите значение около 220-230В.
Вопрос: Как правильно проверить фазу и ноль мультиметром?
Имя: Кирилл
Ответ: Сначала можно найти фазу. Как это сделать, зависит от количества проводов: два или три. В первом случае наконечником алого провода тестера дотрагиваемся до исследуемого проводка. Наконечник темного провода мультиметра прижимаем пальцами или касаемся им заземленного предмета (второй вариант предпочтительнее!). После определения фазы можно найти ноль и заземление.
После определения фазы можно найти ноль и заземление.
Вопрос: Как можно найти фазу в розетке 220В мультиметром?
Имя:
КамильОтвет: Проще всего это сделать, если три проводка: земля, ноль и фаза. Нужно только проверить напряжение всех пар. Между землей и нолём напряжения почти нет, значит, другой проводок — фаза. Если провода два, нужно организовать подходящие условия для движения электричества по прибору.
Вопрос: Как лучше всего найти ноль мультиметром?
Имя: Егор
Ответ: Нужно выключить кабель ввода от заземлительной шины в электрощитке. Когда будет проверяться напряжение между землёй и фазой, 220В не будет, как при проверке ноля и фазы. Если в щитке имеется дифференциальная защитная система, она проявит себя, когда будут проверяться заземлительные проводки относительно иного проводника, даже если он нулевой.
Отвертка индикаторная (фаза 0) 180 мм 6875-304B
- Радиотовары
- Мультиметры, детекторы, измерители
- Отвертка индикаторная (фаза 0) 180 мм 6875-304B

Отвертка индикаторная (фаза 0) 180 мм 6875-304B
- Описание
- Характеристики
- Отзывы (0)
- Доставка
Товар — Отвертка индикаторная (фаза 0) 180 мм 6875-304B производителя НЕ НАЙДЕН знаком многим. Продукция поступила на рынок достаточно давно и уже смогла собрать много положительных отзывов. Полную информацию о характеристиках и способе применения можно получить на официальном сайте производителя. Доставка при самовывозе и отправка по России осуществляется в течении 1-3 рабочих дней.
Продукция поступила на рынок достаточно давно и уже смогла собрать много положительных отзывов. Полную информацию о характеристиках и способе применения можно получить на официальном сайте производителя. Доставка при самовывозе и отправка по России осуществляется в течении 1-3 рабочих дней.
Артикул
277306
НЕ НАЙДЕН
Наименование товара
Отвертка индикаторная (фаза 0) 180 мм 6875-304B
Нет отзывов об этом товаре.
Написать отзыв
Ваше имя:
Достоинства:
Недостатки:
Ваш отзыв
Внимание: HTML не поддерживается! Используйте обычный текст!
Рейтинг
12345
Я прочитал Условия соглашения и согласен с условиями
Наша компания предоставляет несколько вариантов доставки:
Самовывоз: доставка от 1 до 3 рабочих дней. (пункт выдачи г. Чебоксары пр-т. Тракторостроителей дом 72)
(пункт выдачи г. Чебоксары пр-т. Тракторостроителей дом 72)
Доставка по РФ и странам СНГ: Отправка заказа осуществляется в течении 1-3 рабочих дней. Доставка выполняется транспортной компанией Boxberry и CDEK. Сроки доставки заказа расчитывается при оформлении заказа.
Способы оплаты:
- Оплата онлайн на сайте
- Оплата при получении
- Безналичный расчет (для юридических лиц и ИП)
Расчёт стоимости доставки:
Выберите странуАзербайджанАрменияБеларусьГрузияКазахстанКыргызстанМолдоваРоссийская ФедерацияТаджикистанТуркменистанУзбекистанУкраинаНет данных Узнать стоимость доставки
Типы и фазы клинических испытаний
Клинические испытания — это исследования для тестирования новых лекарств, уже одобренных лекарств, устройств или других форм лечения. Во многих клинических испытаниях рассматриваются новые способы обнаружения, диагностики или измерения степени заболевания. Некоторые даже ищут способы предотвратить возникновение болезней. Исследователи до сих пор используют людей-добровольцев для проверки этих методов, и применяются те же правила.
Исследователи до сих пор используют людей-добровольцев для проверки этих методов, и применяются те же правила.
Врачи используют клинические испытания, чтобы узнать, работает ли новое лекарство, лечение или комбинация и безопасно ли их использование для людей. Клинические испытания важны для разработки новых методов лечения серьезных заболеваний, таких как рак. Все новые методы лечения должны пройти клинические испытания, прежде чем они будут одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA). Клинические испытания рака могут занять годы. Могут потребоваться месяцы, если не годы, чтобы увидеть, делает ли лечение рака то, для чего оно предназначено.
Зачем нужны клинические испытания?
Клинические испытания показывают нам, что работает (а что нет) в медицине и здравоохранении. Это лучший способ узнать, что работает при лечении таких заболеваний, как рак. Клинические испытания призваны ответить на несколько важных вопросов:
- Работает ли новое лечение у людей? Если это произойдет, врачи также посмотрят, насколько хорошо это работает.
 Это лучше, чем лечение, используемое сейчас? Если не лучше, то так же хорошо и вызывает меньше побочных эффектов? Или это работает у некоторых людей, которым не помогают современные методы лечения?
Это лучше, чем лечение, используемое сейчас? Если не лучше, то так же хорошо и вызывает меньше побочных эффектов? Или это работает у некоторых людей, которым не помогают современные методы лечения? - Безопасно ли новое лечение? Ни одно лечение или процедура, даже уже широко используемая, не являются безопасными. Но перевешивают ли преимущества нового лечения риски?
- Лучше ли это лечение, чем стандартное лечение этого заболевания? Клинические испытания помогают показать, работает ли новое лекарство или лечение или новая комбинация лечения лучше, чем то, что используется сейчас.
Чтобы ответить на эти вопросы, при одновременном предоставлении как можно меньшему количеству людей неизвестного лечения часто требуется несколько клинических испытаний на разных «фазах». Каждый этап предназначен для того, чтобы ответить на определенные вопросы, сохраняя при этом максимально возможную безопасность участников. Результаты этих фаз показывают, является ли новое лекарство или метод лечения достаточно безопасным и эффективным.
Доклинические (или лабораторные) исследования
Клинические испытания проводятся только после того, как доклинические данные свидетельствуют о том, что новый препарат или метод лечения, вероятно, безопасны и будут работать у людей.
Доклинические исследования, также называемые лабораторными исследованиями, включают:
- Клеточные исследования: часто это первые тесты нового лечения. Чтобы увидеть, может ли это сработать, исследователи изучают влияние нового лечения на раковые клетки, выращенные в лабораторной чашке или пробирке. Эти исследования могут проводиться на раковых клетках человека или раковых клетках животных.
- Исследования на животных: методы лечения, которые кажутся многообещающими в клеточных исследованиях, затем тестируются на раковых заболеваниях у живых животных. Это дает исследователям представление о том, насколько безопасно новое лечение для живых существ.
Доклинические исследования дают много полезной информации, но не всю необходимую. Люди и мыши могут сильно различаться в том, как они поглощают, обрабатывают и избавляются от лекарств или методов лечения. Лечение, которое работает против рака у мышей, может работать или не работать у людей. Также могут быть побочные эффекты и другие проблемы, которые не проявлялись, когда лечение использовалось на мышах, но могли проявляться у людей.
Люди и мыши могут сильно различаться в том, как они поглощают, обрабатывают и избавляются от лекарств или методов лечения. Лечение, которое работает против рака у мышей, может работать или не работать у людей. Также могут быть побочные эффекты и другие проблемы, которые не проявлялись, когда лечение использовалось на мышах, но могли проявляться у людей.
Если доклинические исследования завершены, а лечение все еще кажется многообещающим, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) должно дать разрешение, прежде чем лечение можно будет испытать на людях.
Заявка на получение нового исследуемого препарата (IND)
Прежде чем начать клиническое испытание, оно должно быть одобрено. Заявка или запрос на новый исследуемый препарат или IND должны быть поданы в FDA, если исследователи хотят изучить лекарство на людях. Приложение IND должно содержать определенную информацию, такую как:
- Результаты исследований, чтобы FDA могло решить, безопасно ли лечение для тестирования на людях.

- Как производится препарат, кто его производит, что в нем содержится, насколько он стабилен и многое другое.
- Подробные планы запланированных клинических исследований, называемые протоколами исследований, пересматриваются, чтобы выяснить, могут ли люди подвергаться ненужному риску.
- Подробная информация о группе клинических испытаний, чтобы узнать, обладают ли они знаниями и навыками для проведения клинических испытаний.
Спонсор исследования должен взять на себя обязательство получить информированное согласие от всех участников клинического испытания. Они также должны обеспечить рассмотрение исследования экспертным советом учреждения (IRB) и соблюдение всех правил, необходимых для изучения исследуемых новых препаратов. Каждый этап предназначен для ответа на определенные вопросы. Знание фазы клинического испытания важно, потому что это может дать вам некоторое представление о том, как много известно об изучаемом лечении. Участие в каждой фазе клинического испытания имеет свои преимущества и риски.
Несмотря на то, что проводятся клинические испытания устройств, а также других заболеваний и методов лечения, в описанных здесь примерах фаз клинических испытаний используются лекарства для больных раком.
Фаза 0 клинических испытаний: Изучение того, может ли и как новое лекарство работать
Несмотря на то, что исследования фазы 0 проводятся на людях, этот тип исследований отличается от других фаз клинических испытаний. Цель этого этапа — помочь ускорить и упростить процесс утверждения лекарств. Исследования фазы 0 могут помочь исследователям выяснить, делают ли лекарства то, что от них ожидается. Это может помочь сэкономить время и деньги, которые были бы потрачены на более поздние этапы испытаний.
Исследования фазы 0 используют только несколько небольших доз нового лекарства у нескольких человек. Они могут проверить, достигает ли препарат опухоли, как препарат действует в организме человека и как раковые клетки в организме человека реагируют на препарат. Людям, участвующим в этих исследованиях, могут потребоваться дополнительные анализы, такие как биопсия, сканирование и образцы крови, как часть процесса.
Людям, участвующим в этих исследованиях, могут потребоваться дополнительные анализы, такие как биопсия, сканирование и образцы крови, как часть процесса.
В отличие от других фаз клинических испытаний, практически нет шансов, что люди, участвующие в испытаниях фазы 0, получат пользу. Польза будет для других людей в будущем. А поскольку дозы препаратов низкие, риск для участников исследования также ниже.
Исследования фазы 0 широко не используются, и есть некоторые препараты, для которых они бесполезны. Исследования фазы 0 очень малы, часто в них принимают участие менее 15 человек, и препарат назначают только на короткое время. Они не являются обязательной частью тестирования нового лекарства.
Фаза I клинических испытаний: безопасно ли лечение?
Исследования фазы I нового препарата обычно являются первыми, в которых участвуют люди. Исследования фазы I проводятся, чтобы найти самую высокую дозу нового лечения, которую можно безопасно назначать, не вызывая серьезных побочных эффектов. Хотя лечение было протестировано в лаборатории и на животных, побочные эффекты у людей точно не известны. Эти исследования также помогают принять решение о наилучшем способе проведения нового лечения.
Хотя лечение было протестировано в лаборатории и на животных, побочные эффекты у людей точно не известны. Эти исследования также помогают принять решение о наилучшем способе проведения нового лечения.
Ключевые моменты фазы I клинических испытаний
- Первые несколько человек в исследовании получают очень низкую дозу лечения и находятся под очень пристальным наблюдением. Если есть только незначительные побочные эффекты, следующие несколько участников получают более высокую дозу. Этот процесс продолжается до тех пор, пока врачи не найдут дозу, которая с наибольшей вероятностью подействует, но при этом будет иметь приемлемый уровень побочных эффектов.
- Испытания фазы I также смотрят на то, что лекарство делает с телом и что тело делает с лекарством.
- Безопасность превыше всего. Исследовательская группа внимательно следит за людьми и следит за любыми серьезными побочными эффектами. Из-за небольшого числа людей, участвующих в исследованиях фазы I, редкие побочные эффекты могут не проявляться до более поздних фаз испытаний, когда лечение получает большее количество людей.

- Хотя некоторые люди могут получить пользу от его приема, ответ на заболевание не является основной целью исследования фазы I,
- Плацебо (неактивные препараты) не используются в исследованиях фазы I.
- Испытания фазы I обычно включают небольшое количество людей (до нескольких десятков).
- Испытания фазы I чаще всего включают людей с различными типами рака.
- Эти исследования обычно проводятся в крупных онкологических центрах.
Исследования фазы I несут наибольший потенциальный риск. Но исследования фазы I действительно помогают некоторым пациентам. Для людей с опасными для жизни заболеваниями ключевое значение имеет тщательное взвешивание потенциальных рисков и преимуществ. Иногда люди решают присоединиться к испытаниям фазы I, когда все другие варианты лечения уже опробованы.
Фаза II клинических испытаний: работает ли лечение?
Если новое лечение признано безопасным в ходе клинических испытаний фазы I, проводится клиническое испытание фазы II, чтобы проверить, работает ли оно при определенных типах рака. Польза, которую ищут врачи, зависит от цели лечения. Это может означать, что опухоль уменьшается или исчезает. Или это может означать, что есть длительный период времени, когда рак не становится больше, или есть больше времени, прежде чем рак вернется. В некоторых исследованиях преимуществом может быть улучшение качества жизни. Многие клинические испытания направлены на то, чтобы выяснить, живут ли люди, получающие новое лечение, дольше, чем большинство людей без лечения.
Польза, которую ищут врачи, зависит от цели лечения. Это может означать, что опухоль уменьшается или исчезает. Или это может означать, что есть длительный период времени, когда рак не становится больше, или есть больше времени, прежде чем рак вернется. В некоторых исследованиях преимуществом может быть улучшение качества жизни. Многие клинические испытания направлены на то, чтобы выяснить, живут ли люди, получающие новое лечение, дольше, чем большинство людей без лечения.
Ключевые моменты клинических испытаний фазы II
- Группа из 25-100 пациентов с одним и тем же типом рака получает новое лечение в рамках исследования фазы II. Их лечат с использованием дозы и метода, признанных наиболее безопасными и эффективными в исследованиях фазы I.
- Обычно в клинических испытаниях фазы II все получают одинаковую дозу. Но в некоторых исследованиях фазы II людей случайным образом распределяют по разным группам лечения. Эти группы могут получать разные дозы или получать лечение разными способами, чтобы увидеть, что обеспечивает наилучший баланс безопасности и ответа.
- Плацебо (неактивные препараты) не используются в исследованиях фазы II.
- Исследования фазы II могут проводиться в крупных онкологических центрах, общественных больницах или даже в кабинетах врачей.
Большее количество пациентов получают лечение в ходе испытаний фазы II, поэтому могут наблюдаться менее распространенные побочные эффекты. Если лечение приносит пользу достаточному количеству пациентов, а побочные эффекты не слишком серьезны, начинаются клинические испытания фазы III.
Клинические испытания фазы III: лучше ли это того, что уже доступно?
Лечение, эффективность которого была доказана в клинических испытаниях фазы II, должно пройти еще одну фазу, прежде чем оно будет одобрено для общего применения. Клинические испытания фазы III сравнивают безопасность и эффективность нового лечения с текущим стандартным лечением.
Поскольку врачи еще не знают, какое лечение лучше, участников исследования часто выбирают случайным образом (так называемые рандомизированные ) для получения либо стандартного лечения, либо нового лечения. Когда это возможно, ни врач, ни пациент не знают, какое лечение получает пациент. Этот тип исследования называется двойное слепое исследование . Рандомизация и ослепление обсуждаются более подробно позже.
Когда это возможно, ни врач, ни пациент не знают, какое лечение получает пациент. Этот тип исследования называется двойное слепое исследование . Рандомизация и ослепление обсуждаются более подробно позже.
Ключевые моменты клинических испытаний фазы III
- Большинство клинических испытаний фазы III включают большое количество пациентов, по крайней мере, несколько сотен.
- Эти исследования часто проводятся во многих местах по всей стране (или даже по всему миру) одновременно.
- Клинические испытания фазы III чаще всего проводятся в местных больницах и врачебных кабинетах.
- Эти исследования, как правило, длятся дольше, чем исследования фазы I и II.
- Плацебо могут использоваться в некоторых исследованиях фазы III, но они никогда не используются отдельно, если есть доступное эффективное лечение. Иногда пациенту, которому случайным образом назначают плацебо на часть исследования, в какой-то момент также будет предложено стандартное лечение.

Как и в других исследованиях, за пациентами, участвующими в клинических испытаниях фазы III, внимательно наблюдают на предмет побочных эффектов, и лечение прекращают, если они слишком сложны для лечения.
Подача на утверждение FDA: Заявка на новый лекарственный препарат (NDA)
В Соединенных Штатах, когда клинические испытания фазы III (или иногда испытания фазы II) показывают, что новое лекарство более эффективно или безопасно, чем текущее лечение, подается новая заявка на лекарство. (NDA) подается на утверждение в Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA). FDA рассматривает результаты клинических испытаний и другую соответствующую информацию.
На основании обзора FDA решает, следует ли одобрить лечение для использования у пациентов с заболеванием, на котором тестировалось лекарство. В случае одобрения новое лечение часто становится стандартом лечения, и новые лекарства могут быть проверены на соответствие ему, прежде чем они могут быть одобрены.
Если FDA считает, что необходимы дополнительные доказательства того, что преимущества нового лечения перевешивают его риски, оно может запросить дополнительную информацию или даже потребовать проведения дополнительных исследований.
Клинические испытания фазы IV: что еще нам нужно знать?
Препараты, одобренные FDA, часто находятся под наблюдением в течение длительного периода времени в исследованиях фазы IV. Даже после тестирования нового лекарства на тысячах людей все эффекты лечения могут быть неизвестны. На некоторые вопросы, возможно, еще предстоит ответить. Например, лекарство может получить одобрение FDA, поскольку было показано, что оно снижает риск рецидива рака после лечения. Но значит ли это, что те, у кого он есть, с большей вероятностью проживут дольше? Существуют ли редкие побочные эффекты, которые еще не наблюдались, или побочные эффекты, которые проявляются только после того, как человек принимает препарат в течение длительного времени? На ответы на эти вопросы может уйти еще много лет, и они часто решаются в ходе клинических испытаний IV фазы.
Ключевые моменты клинических испытаний фазы IV
- В исследованиях фазы IV рассматриваются препараты, уже одобренные FDA. Лекарства доступны для врачей, которые могут назначать их пациентам, но для ответа на важные вопросы все еще могут потребоваться исследования фазы IV.
- В этих исследованиях могут участвовать тысячи человек.
- Часто это самый безопасный тип клинических испытаний, потому что лечение уже много изучено и, вероятно, применялось на многих людях. Исследования фазы IV рассматривают безопасность с течением времени.
- В этих исследованиях также могут быть рассмотрены другие аспекты лечения, такие как качество жизни или экономическая эффективность.
Вы можете получить препараты, используемые в исследовании фазы IV, не участвуя в исследовании. И помощь, которую вы получите в исследовании фазы IV, очень похожа на помощь, которую вы могли бы ожидать, если бы вы получали лечение вне испытаний. Но в исследованиях фазы IV вы помогаете исследователям узнать больше о лечении и оказываете услугу будущим пациентам.
Клинические испытания фазы 0: концепции и заблуждения
- Список журналов
- Рукописи авторов HHS
- PMC7185299
Рак Дж. Авторская рукопись; доступно в PMC 2020 27 апреля.
Опубликовано в окончательной редакции как:
Рак J. 2008 май-июнь; 14(3): 133–137.
doi: 10.1097/PPO.0b013e318172d6f3
PMCID: PMC7185299
NIHMSID: NIHMS1575547
PMID: 18536551
, MD, * , PhD, † , PhD, ‡ , PhD, ‡ , MD, * , MD, MS, FACP, † , PhD, ‡ , PhD, † , MBA, PhD, † , PhD, ‡ , DVM, Phd, PhD, ‡ , DVM, Phd, PhD, ‡ , DVM, Phd, PhD, ‡ , DVM, Phd, PhD, ‡ , Phd. , MD, PhD, † , MD, * , PhD, * , PhD, † , PhD, † и , MD * †
, MD, PhD, † , MD, * , PhD, * , PhD, † , PhD, † и , MD * †
Информация об авторе Информация об авторских правах и лицензиях, разработанная в ответах на клинические испытания United Phases
3 Недавнее руководство Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) по исследовательским новым лекарственным средствам (IND) предназначено для ускорения клинической оценки новых молекулярных объектов. Исследовательский IND поддерживает проведение первых испытаний новых исследуемых агентов на людях в субтерапевтических дозах на основе сниженных производственных и токсикологических требований, что позволяет продемонстрировать целевые эффекты лекарственного средства и оценить фармакокинетические-фармакодинамические взаимосвязи у людей на более ранних этапах клинической разработки. Цели фазы 0 клинических испытаний рака состоят в том, чтобы установить как можно раньше — до того, как будет набрано большое количество пациентов и они подверглись потенциальной токсичности, связанной с лекарством, — модулирует ли агент свою мишень в опухоли, и, следовательно, будут ли дальнейшие клинические развитие гарантировано. Здесь мы рассмотрим основные требования к клиническим исследованиям, проводимым в рамках исследовательского IND, и рассмотрим некоторые распространенные заблуждения относительно онкологических исследований фазы 0.
Здесь мы рассмотрим основные требования к клиническим исследованиям, проводимым в рамках исследовательского IND, и рассмотрим некоторые распространенные заблуждения относительно онкологических исследований фазы 0.
Разработка нового противоракового препарата является дорогостоящим, долгосрочным и сопряженным с высоким риском предприятием с частотой неудач более 90%. Более половины новых онкологических препаратов терпят неудачу на более поздних стадиях клинической разработки, что увеличивает затраты и время, необходимые для того, чтобы сделать эффективные методы лечения доступными для пациентов. 1,2 Чтобы ускорить открытие и разработку новых молекулярных соединений, в 2006 г. FDA выпустило руководство по исследовательскому новому лекарству (IND) для поддержки клинической оценки перед повышением дозы, исследованиями безопасности и переносимости, связанными с традиционным IND. 3 Цели и конечные точки исследований фазы 0 (или до фазы I), проводимых в рамках исследовательского IND, могут включать оценку модуляции предполагаемого целевого лекарственного средства у людей; оптимизация методологии анализа мишеней с использованием образцов человека; предоставление фармакокинетических (ФК) данных; оценка ФК/фармакодинамических (ФД) взаимосвязей; и выбор наиболее перспективного ведущего агента из нескольких химических соединений или составов.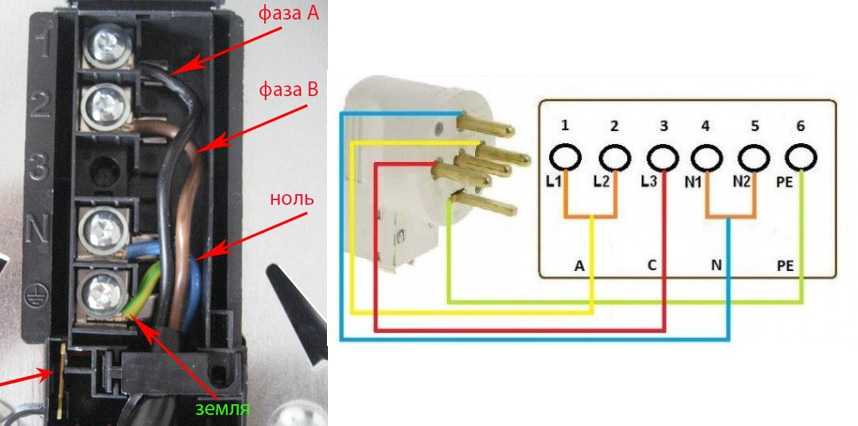 4
4
Основное различие между испытаниями фазы 0 и испытаниями, проводимыми в рамках традиционной IND, заключается в том, что испытания фазы 0 не имеют терапевтической цели. Участникам исследования, которыми могут быть как пациенты, так и здоровые добровольцы, вводят субтерапевтические, но фармакологически активные дозы препарата. Воздействие агента на участников ограничено, но допускается повышение дозы при условии, что конечной точкой не является установление профиля безопасности/токсичности. Поскольку дозы и воздействие лекарств низкие, значительных побочных эффектов, связанных с лекарствами, не ожидается, и FDA разрешает использовать более ограниченные (однократные дозы или короткие курсы) доклинические токсикологические исследования для установления границы безопасности, а не для ограничения дозы. токсичность. Кроме того, из-за небольшого количества исследуемого препарата, необходимого для проведения испытания фазы 0, полномасштабное коммерческое производство клинического уровня надлежащей производственной практики не требуется до начала испытания. Таким образом, испытания фазы 0 могут быть начаты раньше, чем традиционные исследования фазы I, что дает ценную возможность изучить фармакокинетические эффекты и целевые эффекты лекарственного средства у людей намного раньше в клинической разработке агента. Данные, полученные в результате таких пилотных испытаний с участием небольшого числа пациентов, могут служить ориентиром для принятия решений относительно дальнейшей клинической разработки и лучше информировать о дизайне последующих испытаний (2). Данные о ФК и ФД человека помогут ускорить последующие испытания, такие как ускоренные (т. е. с ограниченным уровнем дозы) исследования фазы I, испытания фазы I, сочетающие таргетные агенты с цитотоксическими препаратами, или испытания фазы I/II. На всех этих последующих этапах для продолжения клинической оценки необходимо подать традиционную заявку IND. В 2007 году авторы провели первую фазу 0 клинических испытаний терапевтического агента в онкологии для оценки ABT-888, ингибитора фермента репарации ДНК поли-АДФ-рибозополимеразы у пациентов с запущенными злокачественными новообразованиями.
Таким образом, испытания фазы 0 могут быть начаты раньше, чем традиционные исследования фазы I, что дает ценную возможность изучить фармакокинетические эффекты и целевые эффекты лекарственного средства у людей намного раньше в клинической разработке агента. Данные, полученные в результате таких пилотных испытаний с участием небольшого числа пациентов, могут служить ориентиром для принятия решений относительно дальнейшей клинической разработки и лучше информировать о дизайне последующих испытаний (2). Данные о ФК и ФД человека помогут ускорить последующие испытания, такие как ускоренные (т. е. с ограниченным уровнем дозы) исследования фазы I, испытания фазы I, сочетающие таргетные агенты с цитотоксическими препаратами, или испытания фазы I/II. На всех этих последующих этапах для продолжения клинической оценки необходимо подать традиционную заявку IND. В 2007 году авторы провели первую фазу 0 клинических испытаний терапевтического агента в онкологии для оценки ABT-888, ингибитора фермента репарации ДНК поли-АДФ-рибозополимеразы у пациентов с запущенными злокачественными новообразованиями. 5–7 Потенциальная ценность исследовательского IND для ускорения разработки традиционных лекарств также была признана благодаря его растущему использованию в фармацевтической промышленности. 8 Это особенно полезно для определения приоритетности потенциальных агентов для дальнейшего изучения на самых ранних этапах процесса клинической разработки.
5–7 Потенциальная ценность исследовательского IND для ускорения разработки традиционных лекарств также была признана благодаря его растущему использованию в фармацевтической промышленности. 8 Это особенно полезно для определения приоритетности потенциальных агентов для дальнейшего изучения на самых ранних этапах процесса клинической разработки.
Открыть в отдельном окне
Сокращение сроков клинической разработки с помощью руководства по поисковым новым лекарственным средствам (IND). Проведение испытания фазы 0 в рамках исследовательского IND может сократить время клинической разработки новых агентов и дать информацию для принятия дальнейших клинических решений. A. Испытания фазы I в рамках традиционного IND требуют серьезных доклинических токсикологических исследований и полномасштабного производства исследуемого агента в соответствии с надлежащей производственной практикой перед клиническим применением. Фармакодинамические (ФД) исследования обычно не проводятся до начала испытаний II фазы. B, Фаза 0 испытаний визуализации/биораспределения вводит субфармакологические дозы нового агента пациентам или здоровым добровольцам. Результаты этих испытаний могут быть достаточными для доказательства принципа, и дальнейшие исследования фазы I повышения дозы могут не потребоваться. Эти визуализирующие исследования можно использовать в качестве коррелятивных исследований в последующих исследованиях терапевтических агентов фазы II/III. C, испытания фазы 0 с конечной точкой PD должны иметь валидированный анализ PD до включения в клиническое исследование. Решение о проведении дальнейшей клинической разработки и проведении ускоренных испытаний фазы I/фазы I или испытаний фазы I/II может быть принято на основании того, была ли достигнута цель ФД в испытании фазы 0.
Исследовательское руководство FDA по IND содержит 3 общих примера ранних фаз клинических испытаний, которые касаются (1) ФК или визуализации, (2) фармакологически значимых доз и (3) оценки механизма действия агента. В первом примере, предоставленном FDA, исследования предназначены для получения данных о фармакокинетике, но с использованием доз препаратов, не обладающих фармакологическими эффектами; этот пример вводит понятие «микродозирование». Микродозы определяются в руководстве как менее 1/100 от расчетной в доклинических токсикологических исследованиях на животных для оказания фармакологического эффекта, вплоть до предела 100 мг (или не более 30 нмоль для белковых продуктов). На практике доклинические токсикологические исследования, проведенные в поддержку исследовательского IND, должны продемонстрировать, что доза, в 100 раз превышающая предлагаемую клиническую дозу, не вызывает нежелательных явлений. Для сравнения, начальная доза для первого онкологического исследования на человеке, проводимого в рамках традиционного IND, может составлять 1/10 дозы, которая привела к тяжелой токсичности или смерти у 10% протестированных грызунов. 9 Исследования микродозирования, также называемые скринингом абсорбции, распределения, метаболизма и выделения человека в следовых дозах, включают введение однократной субфармакологической дозы изотопно-меченого препарата для анализа с помощью «сверхчувствительной» ускорительной масс-спектрометрии или позитронно-эмиссионной томографии. Одна из основных проблем, связанных с исследованиями микродоз, заключается в том, что экстраполяция на терапевтические дозы может быть затруднена из-за наличия нелинейной фармакокинетики; в таких обстоятельствах фармакокинетика, определенная с помощью исследования микродоз, не является предиктором фармакокинетики агента при клинических уровнях дозы. 10–12
Важно отличать исследования с применением микродоз от исследований с применением фармакологически активных, но субтерапевтических доз. В первых исследованиях измерялись фармакокинетические параметры лекарственного средства, такие как аффинность связывания и абсорбция, распределение, метаболизм и экскреция. Последний, рассматриваемый во втором и третьем примерах руководства FDA, оценивает конкретные, заранее определенные конечные точки ФК и ФД, представляющие особый интерес для разработки онкологических препаратов. Например, во втором примере руководства FDA исследования фазы 0 фармакологически значимых доз могут установить фармакокинетические параметры (такие как пероральная биодоступность) одного или нескольких исследуемых агентов, оценивая пригодность для дальнейшей разработки в дозах, несущих минимальный риск токсичности, связанной с лекарством. . Начальная доза определяется как 1/50 уровня отсутствия побочных эффектов, определенного в 2-недельном токсикологическом исследовании на грызунах. Если негрызуны являются наиболее чувствительным видом, агент-кандидат должен быть исключен из исследовательского IND. Повышение дозы для желаемого воздействия лекарственного средства или модуляции мишени разрешено, но ограничено в руководстве несколькими критериями максимальной дозы, например дозой, при которой впервые измеряется фармакологический эффект или модуляция мишени, или клинически эквивалентной 1/4 от рекомендуемой дозы. отсутствие наблюдаемого уровня побочных эффектов в 2-недельном токсикологическом исследовании на грызунах или 1/2 площади под кривой наиболее чувствительных видов, в зависимости от того, что меньше.
. Начальная доза определяется как 1/50 уровня отсутствия побочных эффектов, определенного в 2-недельном токсикологическом исследовании на грызунах. Если негрызуны являются наиболее чувствительным видом, агент-кандидат должен быть исключен из исследовательского IND. Повышение дозы для желаемого воздействия лекарственного средства или модуляции мишени разрешено, но ограничено в руководстве несколькими критериями максимальной дозы, например дозой, при которой впервые измеряется фармакологический эффект или модуляция мишени, или клинически эквивалентной 1/4 от рекомендуемой дозы. отсутствие наблюдаемого уровня побочных эффектов в 2-недельном токсикологическом исследовании на грызунах или 1/2 площади под кривой наиболее чувствительных видов, в зависимости от того, что меньше.
Третий пример руководства FDA охватывает исследования фазы 0 для оценки механизма действия агента. Эти исследования включают конечную точку PD, которая отражает активность лекарственного средства, такую как ингибирование целевого фермента в суррогатных образцах или образцах опухолевой ткани. Начальная доза для этих исследований соответствует таковой для исследований, измеряющих конечные точки ФК и ФД, и основана на эффективности на животных моделях. Руководство допускает значительную гибкость в дизайне исследования; в недавнем обзоре описывается, как FDA разрешило фармацевтической компании провести испытание фазы 0 с более длительным периодом дозирования, чем максимальные 7 дней, описанные в руководстве. 8
Начальная доза для этих исследований соответствует таковой для исследований, измеряющих конечные точки ФК и ФД, и основана на эффективности на животных моделях. Руководство допускает значительную гибкость в дизайне исследования; в недавнем обзоре описывается, как FDA разрешило фармацевтической компании провести испытание фазы 0 с более длительным периодом дозирования, чем максимальные 7 дней, описанные в руководстве. 8
Поскольку основное внимание в испытаниях фазы 0 уделяется проверке концепции, а не определению дозы, которую необходимо принять для испытаний фазы II на основе токсичности, необходимое количество участников обычно меньше, чем в испытаниях фазы I. только от 10 до 15. Таким образом, планы исследований фазы 0 должны учитывать статистические ограничения клинических исследований с небольшим размером выборки, аналитические характеристики используемого анализа ФД, вариабельность внутри пациента и молекулярную и гистологическую гетерогенность между пациентами при измерении эффектов ФК/ФД как первичные конечные точки. Вопрос изменчивости внутри пациента вызывает особую озабоченность, когда первичная конечная точка получена из инвазивных биопсий опухоли, которые по своей природе не позволяют часто брать образцы тканей. В этом случае эффекты после лечения должны быть измерены в сравнении с вариабельностью конечной точки до лечения, которую можно исследовать у разных пациентов, а не у отдельного пациента, что существенно затрудняет достижение статистической значимости, поскольку вариабельность конечной точки между пациентами по определению выше (часто гораздо больше), чем внутрибольничная вариабельность.
Вопрос изменчивости внутри пациента вызывает особую озабоченность, когда первичная конечная точка получена из инвазивных биопсий опухоли, которые по своей природе не позволяют часто брать образцы тканей. В этом случае эффекты после лечения должны быть измерены в сравнении с вариабельностью конечной точки до лечения, которую можно исследовать у разных пациентов, а не у отдельного пациента, что существенно затрудняет достижение статистической значимости, поскольку вариабельность конечной точки между пациентами по определению выше (часто гораздо больше), чем внутрибольничная вариабельность.
Право пациентов на участие в онкологических исследованиях фазы 0 и фазы I одинаково в том, что опухоли пациентов, вероятно, будут невосприимчивы к терапии, одобренной FDA; однако исследования фазы 0 из-за их ограниченной продолжительности могут также включать пациентов с вялотекущими заболеваниями, такими как хронический лимфолейкоз или фолликулярные лимфомы, для которых стандартная терапия не показана. Выбор участия в фазе 0, а не в фазе I, проводимой впервые на людях, требует от пациента понимания того, что в случае исследования фазы 0 терапевтический эффект невозможен.
Выбор участия в фазе 0, а не в фазе I, проводимой впервые на людях, требует от пациента понимания того, что в случае исследования фазы 0 терапевтический эффект невозможен.
Решение об оценке нового исследуемого агента в рамках исследовательского, а не традиционного IND зависит от нескольких факторов. Для агента эти факторы включают низкую токсичность и широкий терапевтический индекс на животных моделях, что позволяет продемонстрировать целевую модуляцию при отсутствии значительных побочных эффектов. 4 Для испытаний фазы 0, оценивающих механизм действия, требуется значительный объем ранее существовавшей информации о молекулярной фармакологии лекарственного средства, а также доступность анализа PD, который может надежно измерить действие лекарственного средства на мишень либо непосредственно в опухоли, либо в суррогатном материале. салфетка. Таким образом, одним из препятствий для проведения исследования фазы 0 является наличие ресурсов для разработки анализа, который был бы достаточно чувствительным, устойчивым и надежным для получения значимых результатов в небольшой исследуемой популяции. 13 Анализ также должен быть клинически осуществим в том смысле, что исследуемый целевой эффект можно наблюдать в доступной ткани. Стандартные операционные процедуры обращения с клиническими образцами и их обработки также необходимо оптимизировать в доклинических моделях до начала испытаний. 14 Короче говоря, клиническая квалификация используемого анализа ФД имеет важное значение; анализ должен обеспечивать высокую степень уверенности в том, что эффект лекарственного средства на предполагаемую мишень может быть точно измерен, и должна быть возможность использовать результаты анализа для надежной поддержки решений о клинической разработке.
13 Анализ также должен быть клинически осуществим в том смысле, что исследуемый целевой эффект можно наблюдать в доступной ткани. Стандартные операционные процедуры обращения с клиническими образцами и их обработки также необходимо оптимизировать в доклинических моделях до начала испытаний. 14 Короче говоря, клиническая квалификация используемого анализа ФД имеет важное значение; анализ должен обеспечивать высокую степень уверенности в том, что эффект лекарственного средства на предполагаемую мишень может быть точно измерен, и должна быть возможность использовать результаты анализа для надежной поддержки решений о клинической разработке.
Некоторые из дополнительных проблем, связанных с использованием конечной точки PD в качестве основной цели в исследовании фазы 0, включают небольшое количество участвующих пациентов, внутри- и межпациентную изменчивость выборки опухоли и суррогатной ткани, различную гистологию опухоли в образце клинического исследования. , и молекулярная гетерогенность в пределах типа опухоли, все из которых могут ограничивать возможность демонстрации статистически значимого эффекта PD в опухоли-мишени или суррогатной ткани. 14,15
, и молекулярная гетерогенность в пределах типа опухоли, все из которых могут ограничивать возможность демонстрации статистически значимого эффекта PD в опухоли-мишени или суррогатной ткани. 14,15
Обычная критика испытаний фазы 0 заключается в том, что они представляют собой эксперименты на людях, в частности, на пациентах с неизлечимой формой рака, которые неэтичны, поскольку не предполагают прямой терапевтической пользы. В онкологических исследованиях фазы I и фазы 0 участвуют пациенты с запущенными злокачественными новообразованиями, которые не поддаются стандартной терапии. Этические вопросы, связанные с испытаниями фазы I, в том числе уместность и добровольный характер полученного информированного согласия, научная достоверность исследования, а также восприятие и оценка риска/пользы, стали предметом серьезных дискуссий. 16–19 Испытания фазы 0 еще не подверглись такому же тщательному анализу, но присущее им отсутствие терапевтической цели является очевидной этической проблемой. 20–22
20–22
Как и во всех исследованиях с участием людей, потенциальные риски должны быть тщательно оценены, прежде чем получить одобрение протокола от Институционального наблюдательного совета, и безопасность пациентов имеет первостепенное значение. Институциональный наблюдательный совет должен гарантировать, что, помимо минимизации рисков, «риски для субъектов являются разумными в отношении ожидаемых выгод, если таковые имеются, и важности знаний, которые можно обоснованно ожидать в результате» (45CFR.46.111). 23 Поэтому оценка соотношения потенциального риска и потенциальной пользы при отсутствии прямой пользы для пациентов является сложной задачей. Даже при низких дозах и ограниченных схемах дозирования риски не являются незначительными и включают риски, связанные с процедурами биопсии. По нашему опыту, во время разработки протокола и документа о согласии полезны обсуждения с биоэтиками исследования и связанных с ним рисков. В документе о согласии должно быть четко указано отсутствие терапевтических целей, а также необходимость и связанные с этим риски биопсии опухоли. Кроме того, пациенты должны вербализовать свое понимание этих элементов до подписания документа о согласии. Однако стоит отметить, что пациенты с неизлечимыми заболеваниями ценят информацию как о рисках, связанных с исследованием, так и о ценности знаний, которые можно получить в результате их участия. 24 По опыту авторов, большинство пациентов принимали участие в нескольких клинических испытаниях, прежде чем рассматривать исследование фазы 0, и, таким образом, знакомы с концепциями клинических исследований и исследовательской биопсии. Недавний анализ участников онкологических исследований фазы I не выявил когнитивных, медицинских или демографических факторов, которые могли бы привести к снижению способности принимать обоснованные решения. 25 Кроме того, в отличие от этических соображений, связанных с получением исследовательских биопсий в исследованиях фазы I и II, решение пациента участвовать и предоставить образцы биопсии для исследовательских целей в рамках исследования фазы 0 не омрачено никаким представлением о прямой медицинской пользе.
Кроме того, пациенты должны вербализовать свое понимание этих элементов до подписания документа о согласии. Однако стоит отметить, что пациенты с неизлечимыми заболеваниями ценят информацию как о рисках, связанных с исследованием, так и о ценности знаний, которые можно получить в результате их участия. 24 По опыту авторов, большинство пациентов принимали участие в нескольких клинических испытаниях, прежде чем рассматривать исследование фазы 0, и, таким образом, знакомы с концепциями клинических исследований и исследовательской биопсии. Недавний анализ участников онкологических исследований фазы I не выявил когнитивных, медицинских или демографических факторов, которые могли бы привести к снижению способности принимать обоснованные решения. 25 Кроме того, в отличие от этических соображений, связанных с получением исследовательских биопсий в исследованиях фазы I и II, решение пациента участвовать и предоставить образцы биопсии для исследовательских целей в рамках исследования фазы 0 не омрачено никаким представлением о прямой медицинской пользе. . 26,27 Испытание, основной целью которого является предоставление доказательств воздействия лекарственного средства на предполагаемую цель, не может достичь этой цели без анализа, способного измерить эти эффекты. Поэтому важно убедиться, что существует надежный анализ ФД, который мог бы помочь ответить на научный вопрос с высокой степенью достоверности, прежде чем предлагать пациентам пройти инвазивные процедуры биопсии, которые сопряжены с известными рисками. 26–28
. 26,27 Испытание, основной целью которого является предоставление доказательств воздействия лекарственного средства на предполагаемую цель, не может достичь этой цели без анализа, способного измерить эти эффекты. Поэтому важно убедиться, что существует надежный анализ ФД, который мог бы помочь ответить на научный вопрос с высокой степенью достоверности, прежде чем предлагать пациентам пройти инвазивные процедуры биопсии, которые сопряжены с известными рисками. 26–28
Желание пациентов участвовать в исследовании, предназначенном исключительно для получения общих знаний, примечательно и проистекает из желания помочь будущим больным раком. Поэтому важно информировать пациентов о результатах исследований и о том, как они повлияли на дальнейшее развитие возбудителя. Также важно обеспечить, чтобы участие в испытании фазы 0 не задерживает и не исключает пациентов из участия в других клинических испытаниях, которые действительно предлагают возможность прямой пользы. Этого можно достичь путем ограничения периода вымывания из предшествующей терапии (например, не более 2 недель) как для включения в исследование фазы 0, так и после завершения исследования перед включением в другое исследование. Кроме того, участие в исследовании фазы 0 не должно исключать пациентов из участия в последующих, более поздних стадиях исследования этого агента; теперь в нескольких протоколах фазы I и II NCI есть общий язык, специально посвященный этой проблеме. Мы надеемся, что этот язык будет широко принят другими центрами исследования рака.
Этого можно достичь путем ограничения периода вымывания из предшествующей терапии (например, не более 2 недель) как для включения в исследование фазы 0, так и после завершения исследования перед включением в другое исследование. Кроме того, участие в исследовании фазы 0 не должно исключать пациентов из участия в последующих, более поздних стадиях исследования этого агента; теперь в нескольких протоколах фазы I и II NCI есть общий язык, специально посвященный этой проблеме. Мы надеемся, что этот язык будет широко принят другими центрами исследования рака.
Испытания фазы 0 могут помочь решить некоторые из наиболее сложных проблем разработки новых лекарств в онкологии, помогая определить приоритетность потенциальных агентов для будущих исследований, сокращая сроки разработки и демонстрируя экспериментальное ингибирование мишени. Например, результатов исследования с визуализацией фазы 0 может быть достаточно, чтобы установить доказательство принципа и устранить необходимость в испытании фазы I с повышением дозы; визуализация вместо этого может быть включена в качестве корреляционного исследования в последующие оценки терапевтических агентов фазы II/III. Испытание фазы 0 с конечной точкой PD, которая соответствует его цели, может поддержать решение о переходе к ускоренной фазе I, комбинированной фазе I или испытаниям фазы I/II. Важно подчеркнуть, что испытания фазы 0 не заменят испытания фазы I, проводимые в рамках традиционной IND для установления максимально переносимой дозы и профиля токсичности лекарственного средства. Не все исследуемые агенты подходят для оценки фазы 0. Значительные затраты времени и ресурсов необходимы для разработки подходящих анализов ФД и процедур обращения с образцами. Кроме того, у исследователей могут возникнуть трудности с разработкой ресурсов для исследований фазы 0, поскольку нетерапевтические клинические испытания не покрываются большинством сторонних плательщиков. Этические соображения, необходимые для проведения испытания фазы 0, не являются незначительными, но откровенное и открытое обсуждение с участниками до, во время и после испытания будет взаимовыгодным.
Испытание фазы 0 с конечной точкой PD, которая соответствует его цели, может поддержать решение о переходе к ускоренной фазе I, комбинированной фазе I или испытаниям фазы I/II. Важно подчеркнуть, что испытания фазы 0 не заменят испытания фазы I, проводимые в рамках традиционной IND для установления максимально переносимой дозы и профиля токсичности лекарственного средства. Не все исследуемые агенты подходят для оценки фазы 0. Значительные затраты времени и ресурсов необходимы для разработки подходящих анализов ФД и процедур обращения с образцами. Кроме того, у исследователей могут возникнуть трудности с разработкой ресурсов для исследований фазы 0, поскольку нетерапевтические клинические испытания не покрываются большинством сторонних плательщиков. Этические соображения, необходимые для проведения испытания фазы 0, не являются незначительными, но откровенное и открытое обсуждение с участниками до, во время и после испытания будет взаимовыгодным.
Испытания фазы 0 предлагают возможность оценить фармакокинетику и подтвердить влияние агента на предполагаемую молекулярную мишень в человеческих образцах на гораздо более ранних этапах клинической разработки. На сегодняшний день опыт исследований, проведенных в рамках поисковых IND, ограничен, но положителен. 8 Если действие лекарственного средства на мишень можно оценить на более ранних этапах цикла разработки лекарственного средства и требуется меньше пациентов, чем при традиционном IND, то клинические испытания будут меньше, а сроки разработки могут быть сжаты.
На сегодняшний день опыт исследований, проведенных в рамках поисковых IND, ограничен, но положителен. 8 Если действие лекарственного средства на мишень можно оценить на более ранних этапах цикла разработки лекарственного средства и требуется меньше пациентов, чем при традиционном IND, то клинические испытания будут меньше, а сроки разработки могут быть сжаты.
Авторы благодарят д-ра Гуилио Драэтту за вдохновение и г-жу Джину Уленбраук, SAIC-Frederick, Inc., за редакторскую помощь в подготовке этой рукописи.
Этот проект полностью или частично финансируется за счет федеральных средств Национального института рака, Национальных институтов здравоохранения по контракту N01-CO-12400.
Содержание этой публикации не обязательно отражает точку зрения или политику Министерства здравоохранения и социальных служб, а упоминание торговых наименований, коммерческих продуктов или организаций не означает их одобрения правительством США.
1. Кола И., Лэндис Дж. Может ли фармацевтическая промышленность снизить процент отсева?
Nat Rev Drug Discov. 2004; 3: 711–715. [PubMed] [Google Scholar]
Может ли фармацевтическая промышленность снизить процент отсева?
Nat Rev Drug Discov. 2004; 3: 711–715. [PubMed] [Google Scholar]
2. Министерство здравоохранения и социальных служб США, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов. Инновации или стагнация? Проблемы и возможности на критическом пути к новым медицинским продуктам. Маршировать 2004. Доступно по адресу: http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.pdf. Доступ 21 февраля 2008 г.
3. Министерство здравоохранения и социальных служб США, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов. Руководство для промышленности, исследователей и обозревателей: исследовательские исследования IND. январь 2006. Доступно по адресу: http://www.fda.gov/cder/guidance/7086fnl.pdf. Доступ 21 февраля 2008 г.
4. Куммар С., Киндерс Р., Рубинштейн Л. и соавт.
Сокращение сроков разработки лекарств в онкологии с использованием нулевой фазы испытаний. Нат Рев Рак. 2007; 7: 131–139. [PubMed] [Google Scholar]
Нат Рев Рак. 2007; 7: 131–139. [PubMed] [Google Scholar]
5. Kummar S, Kinders R, Gutierrez M, et al. Ингибирование поли(АДФ-рибозо)полимеразы (PARP) с помощью ABT-888 у пациентов с запущенными злокачественными новообразованиями: результаты исследования фазы 0 [аннотация]. Дж. Клин Онкол. 2007 г.; 25:3518. [Google Scholar]
6. Donawho CK, Luo Y, Luo Y, et al. ABT-888, перорально активный ингибитор поли(АДФ-рибозы) полимеразы, который потенцирует ДНК-повреждающие агенты в доклинических моделях опухолей. Клин Рак Рез. 2007; 13: 2728–2737. [PubMed] [Академия Google]
7. Ратнам К., Лоу Дж.А. Современные разработки клинических ингибиторов поли(АДФ-рибозо)полимеразы в онкологии. Клин Рак Рез. 2007; 13: 1383–1388. [PubMed] [Google Scholar]
8. Robinson WT. Инновационные подходы к регулированию раннего развития: expIND, expCTA, микродозирование. Клин Фармакол Тер. 2008; 83: 358–360. [PubMed] [Google Scholar]
9. DeGeorge JJ, Ahn CH, Andrews PA, et al.
Нормативные аспекты доклинической разработки противоопухолевых препаратов. Рак Chemother Pharmacol. 1998;41:173–185. [PubMed] [Google Scholar]
Рак Chemother Pharmacol. 1998;41:173–185. [PubMed] [Google Scholar]
10. Лаппин Г., Гарнер Р.С. Большая физика, малые дозы: использование АМС и ПЭТ для микродозирования лекарств для разработки человеком. Nat Rev Drug Discov. 2003; 2: 233–240. [PubMed] [Google Scholar]
11. Lappin G, Kuhnz W, Jochemsen R, et al. Использование микродозирования для прогнозирования фармакокинетики в терапевтической дозе: опыт применения 5 препаратов. Клин Фармакол Тер. 2006; 80: 203–215. [PubMed] [Google Scholar]
12. Бойд Р.А., Лалонд Р.Л. Нетрадиционные подходы к первым исследованиям на людях для повышения эффективности разработки лекарств: окажут ли значительное влияние исследования микродоз? Клин Фармакол Тер. 2007; 81: 24–26. [PubMed] [Академия Google]
13. Этапы разработки фармакодинамических тестов.
Веб-сайт программы развивающей терапии Национального института рака. Доступно по адресу: http://dtp.nci.nih.gov/docs/phase0/PharmacoDynamicAssaydeveloment.html.
Доступ
21 февраля 2008 г.
14. Kinders RJ, Hollingshead M, Parchment RE, et al. Доклиническое моделирование протокола клинического исследования фазы 0 [аннотация]. Дж. Клин Онкол. 2007;25: 14058. [Google Scholar]
15. Бетенский Р.А., Луис Д.Н., Кэрнкросс Дж.Г. и др. Влияние непризнанной молекулярной гетерогенности на рандомизированные клинические испытания. Дж. Клин Онкол. 2002;20:2495–2499. [PubMed] [Google Scholar]
16. Emanuel EJ, Wendler D, Grady C. Что делает клинические исследования этичными? ДЖАМА. 2000; 283:2701–2711. [PubMed] [Google Scholar]
17. Агравал М., Эмануэль Э.Дж. Этика онкологических исследований фазы I: пересмотр аргументов и данных. ДЖАМА. 2003; 290:1075–1082. [PubMed] [Google Scholar]
18. Joffe S, Miller FG. Переосмысление оценки риска и пользы для исследований рака I фазы. Дж. Клин Онкол. 2006; 24:2987–2990. [PubMed] [Академия Google]
19. Koyfman SA, Agrawal M, Garrett-Mayer E, et al.
Риски и польза, связанные с новыми дизайнами онкологических исследований фазы 1. Рак. 2007; 110: 1115–1124. [PubMed] [Google Scholar]
Рак. 2007; 110: 1115–1124. [PubMed] [Google Scholar]
20. Киммельман Дж. Этика на этапе 0: прояснение вопросов. J Law Med Ethics. 2007; 35: 727–733. [PubMed] [Google Scholar]
21. Hill TP. Испытания фазы 0: являются ли они этически сложными? Клин Рак Рез. 2007; 13: 783–784. [PubMed] [Google Scholar]
22. Marchetti S, Schellens JHM. Влияние рекомендаций FDA и EMEA на разработку лекарств в отношении испытаний фазы 0. Бр Дж Рак. 2007 г.; 97: 577–581. [Бесплатная статья PMC] [PubMed] [Google Scholar]
23. Министерство здравоохранения и социальных служб США. Свод федеральных правил, раздел 45 — Общественное благосостояние, Министерство здравоохранения и социальных служб, часть 46: Защита людей. Пересмотрено Июнь 23, 2005. Доступно по адресу: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm. Доступ 21 февраля 2008 г.
24. Агравал М. Добровольность клинических исследований в конце жизни. J Управление симптомами боли. 2003; 25:S25–S32. [PubMed] [Академия Google]
25.